Tutorial
These pages describe how to use DENSS most effectively to reconstruct your particle from your solution scattering data. For additional instructions for visualizing and assessing the results, visit the Tips page. There’s also a new Video Tutorial to show you how to download, install, and run DENSS step-by-step.
Background
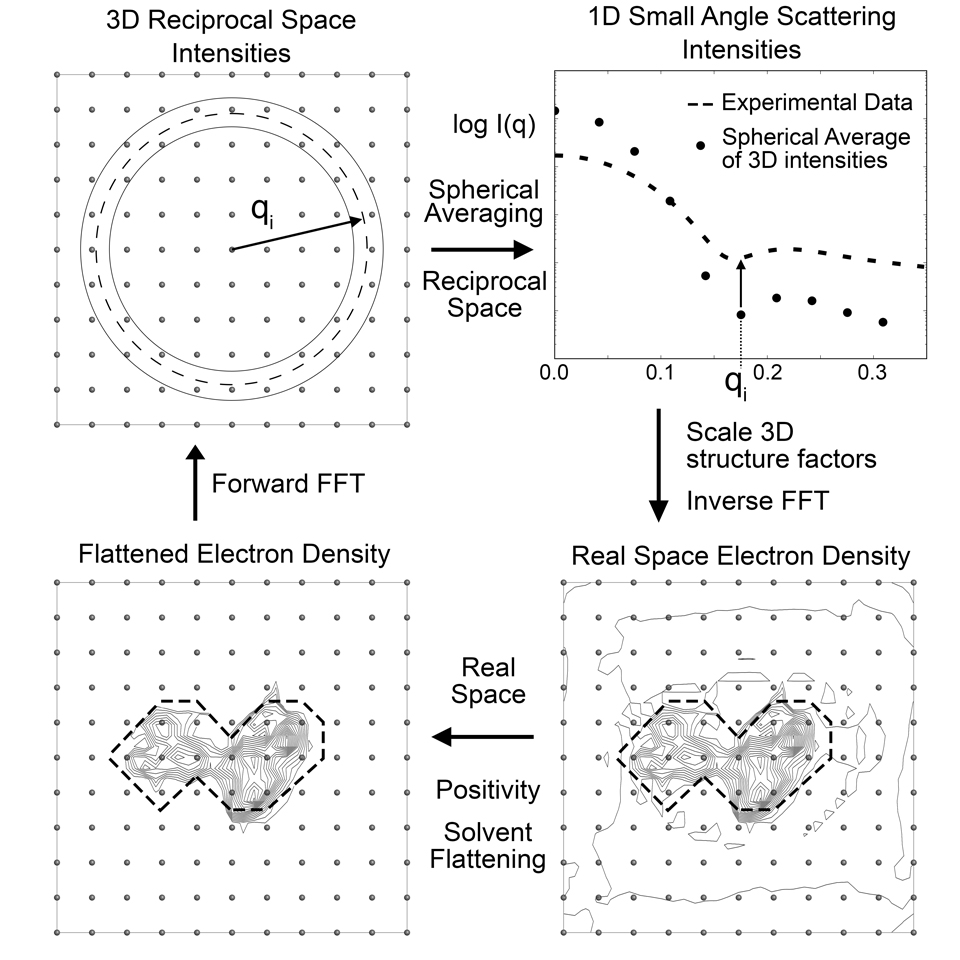
To understand how to run DENSS and how to use all of its options most appropriately, it helps to understand how DENSS works. DENSS is an algorithm used for calculating ab initio electron density maps directly from solution scattering data. DENSS implements a novel iterative structure factor retrieval algorithm to cycle between real space density and reciprocal space structure factors, applying appropriate restraints in each domain to obtain a set of structure factors whose intensities are consistent with experimental data and whose electron density is consistent with expected real space properties of particles.
DENSS utilizes the NumPy Fast Fourier Transform for moving between real and reciprocal space domains. Each domain is represented by a grid of points (Cartesian), N x N x N. N (the number of samples in one dimension) is determined by the size of the system and the desired resolution. The real space size of the box is determined by the maximum dimension of the particle, D, and the desired sampling ratio. Larger sampling ratio results in a larger real space box and therefore a higher sampling in reciprocal space (i.e. distance between data points in q). Smaller voxel size in real space corresponds to higher spatial resolution and therefore to a larger maximum q value in reciprocal space.
The reciprocal space restraints are pretty straight forward, i.e. the data. The real space restraints are imposed by defining a “support”, i.e. the region of space (which voxels) are allowed to have density. Outside of this support the density is set to zero, inside the support the density is required to be positive and real valued. The selection of which voxels are part of the support is regularly updated throughout the reconstruction and is determined by the shrinkwrap algorithm.
Questions?
If you have any questions about any of these options or more generally how to run DENSS, please send an email using the contact form below.